
Medical Device Dangers: A KVUE Special Broadcast
About one in 10 Americans, will have some kind of medical device implanted in their bodies. These devices do not undergo the same scrutiny as pharmaceuticals.
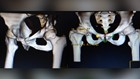
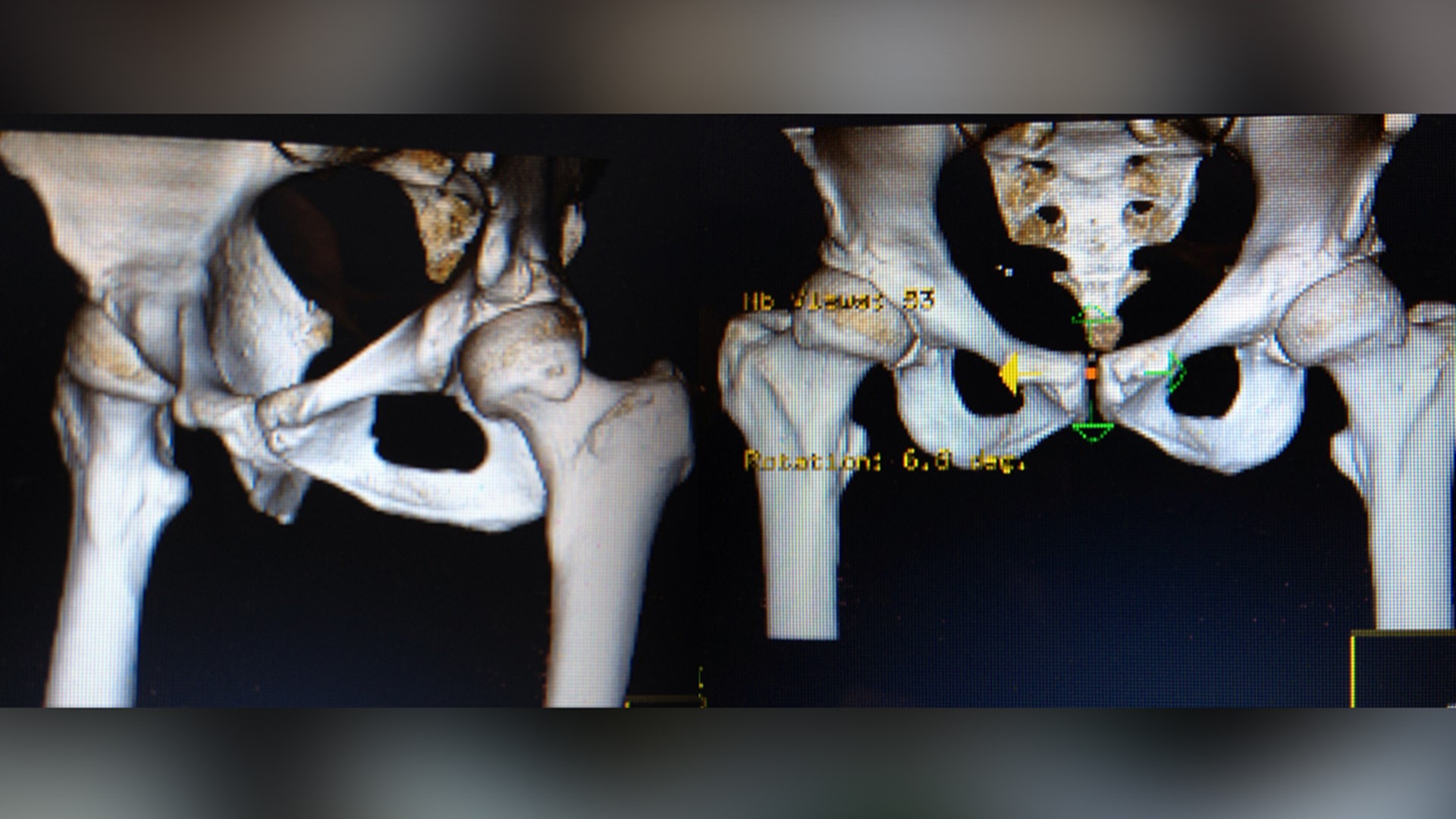
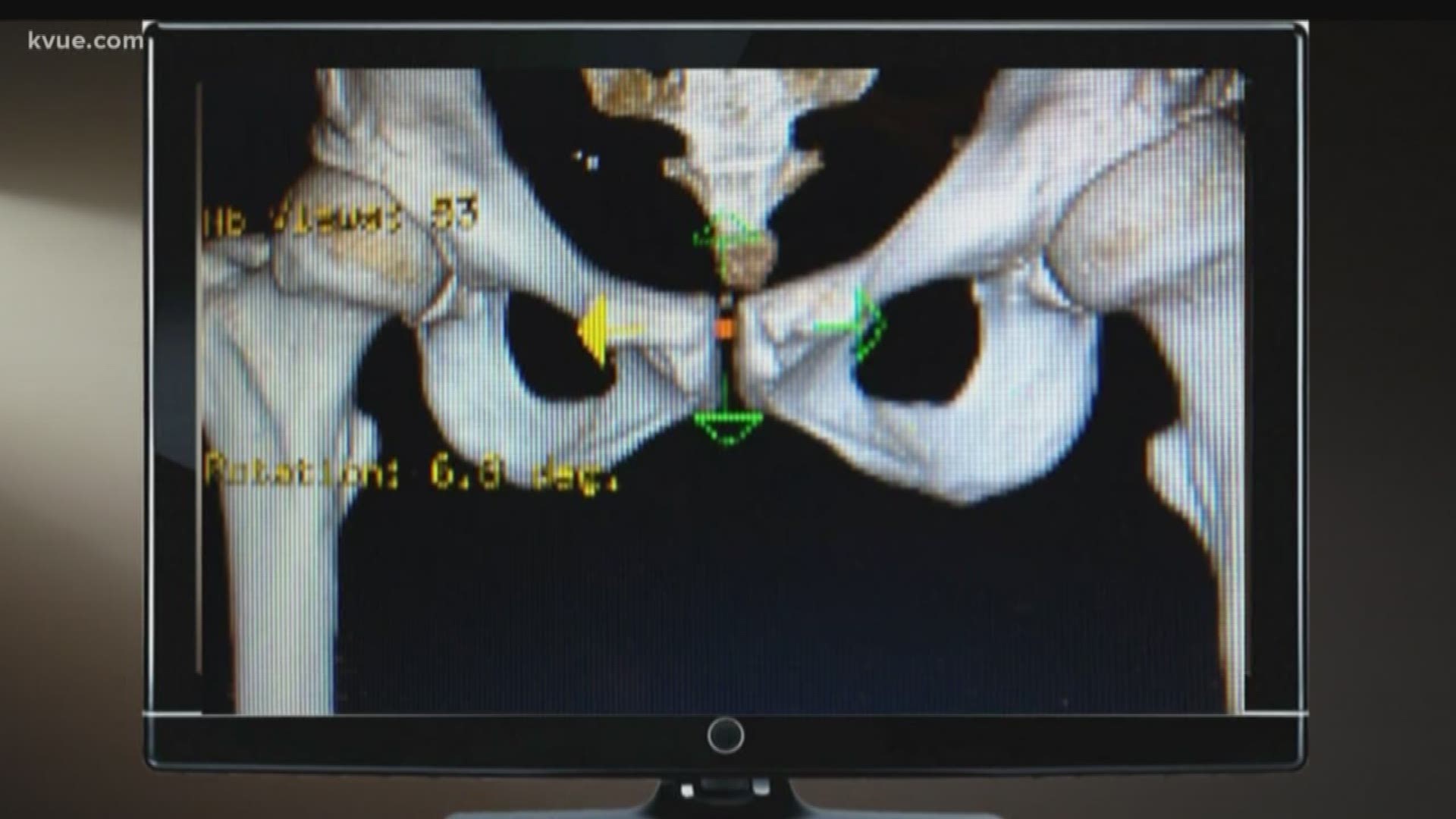
About 32 million Americans, one in 10, will have some kind of medical device implanted in their bodies. A joint, a pump, a piece of wire or mesh.
We expect that these devices undergo rigorous testing and approval. But the KVUE Defenders discovered the FDA does not require the same standards for medical devices as it does prescription drugs.
Metal-on-metal hip implant issues Frances Scott's story
The last 10 years of Frances Scott’s life has left her teetering between loss and pain. Sharing this part of her life is not easy.
“I stayed in shape. I ran, I did everything I was supposed to do,” Scott said in her backyard as tears streamed down her face. “I had to do this twice. There are dishes that I can't stand to do. There's laundry on the floor that I can't bend to pick up. I was going to be a good mom. I was going to take them skiing. I was going to play basketball with them. They took all that from me.”
RELATED:
Once a successful television anchor in North Carolina, she was an active mom in her 30s with a husband and three kids. They were building a good life.
Hip pain then took over.
“[Doctors] said, ‘We think you were born without sockets and when you stood up to walk, the sockets formed in the wrong place,’” she recalled.

A state-of-the-art device leads to even more pain Doctors recommend a double hip replacement
“So, it’s like if you have bad alignment on your car and you drive a long way – you’re going to wear out the tires faster,” she said. “And I had been a runner, an athlete, very active. I'm not a sit still kind of person. So I had put high miles on my hips.”
She said her surgeon suggested the Pinnacle with Ultamet liner: A state-of-the-art, metal-on-metal joint made by Johnson and Johnson Depuy.
It was a major surgery for a 39-year-old mom, but doctors said she’d be back on her feet in a few months.
"I just never got out of pain," she said.
It took a toll on her family.
“I would say no part of it was normal,” said Skip Phillips, her husband.
Her condition got worse.
“I was trying to go back to work and I had all these oozing boils all over my face. It was really upsetting. That's when I started noticing the tinnitus, the hearing loss,” she said.
And the memory loss.
“I couldn't figure out how to get back to work. It was terrifying,” she said. “I would notice my hands would shake.”
On their website, the FDA warns patients of the symptoms of metal-on-metal hip devices:
- General hypersensitivity reaction (skin rash)
- Cardiomyopathy
- Neurological changes including sensory changes (auditory, or visual impairments)
- Psychological status change (including depression or cognitive impairment)
- Renal function impairment
- Thyroid dysfunction (including neck discomfort, fatigue, weight gain or feeling cold.
Searching for answers 'I can find out more about my child's car seat than I can find out about what's implanted in my body'
Frustrated, she searched for answers. But in the U.S., there is no medical device registry.
“I can find out more about my child's car seat than I can find out about what's implanted in my body,” said Scott.
She did find research from England that showed patients with the same implant were suffering from metal poisoning.
"I kept wondering, but everyone I saw kept saying, ‘No. The Pinnacle [with Ultamet liner] is great. It’s a great hip. Nobody else is having problems. Your X-rays look good,’” Scott said.
She began to feel helpless.
"They kept saying, ‘I don't know why you keep coming back because we can't help you.’ Then they stopped taking my appointments and I knew I was on my own," she recalled.
She lost her job, had to sell her home and moved her family to Texas.
“It's a tragedy,” said Mike Lanier, an attorney who does not represent Frances, but has successfully represented hundreds of people against the makers of metal hip replacements, including the Pinnacle with Ultamet liner and Johnson and Johnson Depuy.
"I’ve represented hundreds of people who have had this defective implant in them and the experiences have had a wide range,” he said. “Some people have experienced everything from hearing loss or systemic loss as those metal ions or metal debris moves throughout the body. Some have experienced heart issues. Some have experienced memory issues. Most experience pain around the implant and a loss of muscle and ligaments.”
The head of Johnson and Johnson Depuy's development team warned company leaders of issues with metal-on-metal implants in 1997 before patients started getting Pinnacle with Ultamet liner implants.
A memo filed in court shows "wear is a major problem" and metal-on-metal was a "bad engineering” design. Click here to read the memo.
And in simulator testing for the device, “performance is unpredictable.” The device is shown to work well for period of time before “suffering a sudden catastrophic breakdown,” which includes releasing “a large volume of wear debris.”
“The makers of this had full warning from their development team that this was not a product that was safe to put into people,” said Lanier.
How the FDA approves medical devices 'It's a tragedy'
The FDA sent KVUE a statement saying in part that it is working to modernize the way new medical devices come to market and improve patient safety and performance.
Medical devices are approved through two different processes: 510(k) or petition for premarket submission (PMA).
The majority of medical devices are approved through the 510(k) process.
“This is the fastest and most economical way to get your medical device cleared. FDA guidelines state that a 510(k) should be submitted at least 90 days before the device is marketed,” the FDA said.
If a product is “completely new to the market or classified as a ‘Class III device,’ you must apply for a PMA.
“When there isn’t enough existing research to prove the safety and effectiveness of your device or it’s deemed high risk, the FDA requires extensive scientific evidence to determine that it is ready to be marketed. This generally includes both laboratory and clinical trial data,” the FDA said.
The FDA oversees all medical devices and drugs but the approval process for both is very different. Drugs have to undergo human trials. Most medical devices do not. Click here for a breakdown of how to FDA looks at medical devices.
“So many of the doctors that we've put under oath about this were stunned to find out that the company never tested the device in people,” said Lanier. “The company tested the device in little machines called ‘simulators’ that did not simulate true human activity and never tested the effect that the debris would have on the human body. It's a tragedy."
"I never even thought that something could be implanted in me that had never been tested in a human being,” said Frances.
A known problem at the FDA 'A game of whack-a-mole'
“Essentially, we're just playing a game of whack-a-mole,” said FDA Director Dr. Jeff Shuren in a video from 2015.
The FDA has known for years that the approval-process for medical devices is a problem.
“One device manufacturer can meet FDA requirements and still make a poor quality device,” Shuren said.
Lanier said thousands of patients are paying the price.
"I think a lot of people assume the FDA tests devices,” Lanier said. “And when they find out the FDA does not that the medical device industry, by and large, is a self-policing industry that exists to make profit, then they begin to understand there are issues.”
A growing number of suits Millions paid in attorney general claims
Johnson and Johnson Depuy faces more than 10,000 lawsuits across the United States.
On Jan. 22, 2019, the company agreed to pay $120 million to resolve state attorneys general claims of deceptive marketing in metal-on-metal hip implants. Some of that money – $8.5 million – stayed in Texas.
Those devices were pulled off the market in August 2013, two years after Scott got her metal-on-metal hips.
"I get really angry about this,” said Lanier. “There's no call for putting profits over safety. This is the 21st century. This is the United States of America. We should know better.”
"We were without our own consent made to be guinea pigs," said Scott.
Her new normal Scott finds help
Seven years after her first surgery, Scott finally found a doctor to remove her metal hips.
“I would say about 80% of the symptoms that were really troubling me went away within weeks,” she said.
But she said she still suffers from hearing and vision loss. And because the metal destroyed some of her tissue, she dislocated a hip last summer.
Frances has joined the 10,000-plus others who have filed suit against Johnson and Johnson Depuy.
“I'm not the worst case,” she said.
For Frances’ family, time in her 40s is much different than she imagined.
Her pain persists. By afternoon she is too often forced to sit.
"What I grieve most you can't give me back,” said Frances.
So she presses on with a greater purpose.
“I want this fixed. I want the laws changed. I want this changed so that this can't happen to other people,” she said.
We reached out to Johnson and Johnson Depuy. Here’s what the company said:
“We have no greater responsibility than to the patients who use our products, and we are confident the evidence presented at trial will show that the company acted appropriately and responsibly in the design and testing of ULTAMET® Metal-on-Metal. ULTAMET Metal-on-Metal is backed by a strong track record of clinical data showing reduced pain and restored mobility for patients suffering from chronic hip pain. We remain committed to the long-term defense of the allegations in these lawsuits."
FDA changes rules for metal-on-metal hips
There is still no medical device registry, meaning no one place that stores the history of devices exists.
When a device is removed from a person for medical reasons, that device is sent back to the manufacturer – not the federal government – to figure out why it failed.
The FDA now has stricter requirements for metal-on-metal hips, but there are currently no FDA-approved metal-on-metal hips on the market.
More victims of hip implants Meet Kari Light
When doctors recommend surgery, especially for a medical device, you expect to be able to find out the risks and potential side effects. But finding information about the potential dangers of medical devices is difficult if not impossible.
Working from home is a necessity for 54-year-old Kari Light.
“I can't walk from here to the door without them grinding and trying to come out of the socket,” said Light.
A hip deformity left her in a wheelchair until doctors recommended a double hip replacement in 2014.
“I thought everything was going to be better after that and it was, for a while,” she recalled.
Within six months, the pain and clicking became too much. She went back to the doctor.
“He's like, 'I looked at your X-rays and everything looks good. Your components are perfect,'” she said.
For years, she suffered in silence, living with the pain.
“I've missed so many school programs of my grandkids because I either couldn't walk or I felt so sick that I couldn't get out of bed,” said Light.
Seeing Frances Scott talk about her struggle with a double hip replacement, a device she said was making her sick, hit home.
“I was just shocked,” said Light. “I was like, 'Oh my God, that's me.'”
The KVUE Defenders showed how the FDA has lax testing requirements and little regulation over implanted medical devices. We also uncovered more than 27,000 reports of injuries from hip implants and 111 deaths. But you won't find that information on the FDA's website.
RELATED:
“I think we need legislation to mandate a registry,” said Texas Congressman Lloyd Doggett.
He hopes to change that by sponsoring the Medical Device Safety Act, which would create a medical device registry.
“We are trying to draw that balance between providing and encouraging innovation but protecting the consumer, and I don't think we've drawn it very well in this area,” Doggett said.
“I'm not the worst case,” said Scott.
Dozens of people from across the country are now sharing their stories with Frances, Representative Doggett, and the KVUE Defenders. People like Light, who just this month suffered a seizure.
It was enough for doctors to finally order a test to check the metal ions in her blood.
Although grateful, she said she doesn't need a test to know something is not right.
“I can't walk my dog down to the dog park five houses down, I have to drive. I can't stand for more than two or three minutes without being in excruciating pain,” she said.
Her search for answers continues and now she hopes the FDA takes action.
“I don't understand why they keep turning their face away from these very blatant and increasing number of cases of people who suffer horribly,” said Light.
Medical device makers apologize Women injured by transvaginal message
Nearly one in every five women suffer from prolapse, a condition where pelvic muscles no longer support pelvic organs.
For years, surgeons made repairs using tissue from women's bodies. But, in recent years, a surgical mesh has been used to treat prolapse.
Tuesday, following thousands of complaints and years of warnings from the FDA itself, a public hearing shed light on what is growing evidence of medical device dangers.
“This is affecting lives and it’s major and no one is really listening to us,” said Barbara Melling, a patient who is suffering from complications of surgical mesh.
Women from across the country gathered in Maryland to share their stories about how mesh products changed their lives.
“If there are 100,000 women injured by mesh for any reason it should not be on the market? If tomatoes are killing people they take them off the market,” said Lana Keeton, who had a sling implanted in her.
“I am one of over 100,000 women who have been severely and permanently harmed by the devastating complications from the interior transvaginal mesh,” said Jodie Callahan.
RELATED:
Callahan is a mother who had surgery in 2010 at the age of 45.
“This changed my life and made me limit a lot of activities with my family. I went to nine different specialists – all trying to help me deal with this,” she recalled.
Three years later, after years of pain, a surgeon removed the mesh.
“During the procedure, I experienced life-threatening complications due to the extensive lengths to remove the mesh," she said. "The doctor reported the mesh had eroded into my bladder, attached to my bowels, my nerves and my muscles and the incontinence sling had twisted my urethra, so voiding [emptying her bladder] was nearly impossible. They also removed a large gelatinous cyst that was in my cervix that was engulfed in mesh.”
Last month she learned she has to undergo another surgery. She said her doctor found more plastic mesh inside her body.
“I am scared, frustrated and devastated that I will have to do this again,” she cried.
Medical device makers apologized to the women.
“Boston scientific is purposeful and committed to provide medical devices that are safe and effective and supported by clinical evidence supported by physician training and the best possible outcomes,” he said, citing studies that show the mesh devices continue to improve.
“On behalf of Boston Scientific, and myself as a physician, I want to express my deepest sympathy and my sincere empathy for those women who have been harmed by surgical mesh therapies,” said Dr. Ronald Morton Jr., a medical doctor with the urology division at Boston Scientific.
The Defenders have learned the FDA warned about serious complications involving mesh implants in 2008. In 2011, the FDA mandated companies produce studies to show the safety of the devices.
"In 2011 Public Citizen and I petitioned the FDA to ban and label all mesh products for vaginal repair of POP [pelving organ prolapse] because these devices offer no clinically proven benefits in comparison with non-mesh repair of POP and have high rates of serious complications. The FDA denied our petition in 2014. Surgical mesh for transvaginal POP repair is a quintessential example of the deficiencies of the FDA’s oversight of medical devices, particularly those that are permanently implanted,” said Dr. Michael Carome, M.D. of Public Citizen.
“I truly hope the FDA is serious about addressing the research, development, training as well as adequate oversight for future medical devices,” said Callahan.
The FDA will continue to look at the evidence presented to determine if more action should be taken for surgical mesh.
U.S. Congressman Lloyd Doggett is sponsoring the Medical Device Safety Act as a result of our original investigation and sent us this statement:
“The danger is not limited to one medical device, one manufacturer or one lawsuit," said Congressman Doggett. "The KVUE Defenders has done a really good job reporting this and the urgent need to get some action, transparency and accountability and empower patients and help them not to have to endure the same needless suffering. Mindful of that is what I'm doing is supporting an improved medical device registry and legislation to help patients harmed by medical devices seek damages from the device manufacturer.”
Hernia mesh complications Thousands of lawsuits
Stuck at home is not how Carmen Pacheco expected to live out life in her 50s.
“I've been in and out of the hospital about 60, 70 times since I've had this,” she said.
Her pain and worry matched by her mountain of medical bills. A spiral that began six years ago when doctors performed what most consider routine surgeries, implanting plastic mesh for pelvic prolapse and a hernia.
“They never told me what could happen and a year to the day I had a strangulated hernia. The ambulance had to come pick me up,” said Pacheco.
A second surgery resulted in a massive infection, followed by a third surgery and more pain.
“I can't do nothing now,” she said. “I can't even eat.”
“I've seen a lot of plaintiffs have a lifetime of injuries as a result, a lifetime of additional surgeries in pursuit of repairing what's been done,” said Melissa Nafash, who does not represent Pacheco but is a New York attorney whose firm has more than 10,000 hernia mesh cases.
“Due to design defects in the meshes, often there is a surgery to remove that mesh. Polypropylene, which a lot of these products are made of, can be thought of like a window screen, and tissue is intended to grow into that screen. It's as if you pour cement through a screen and let it dry and try to pull that screen out. It has become a part of the body,” she said.

Sound familiar? It should.
In February, dozens of women shared their stories at an FDA hearing about the problems associated with pelvic mesh.
“The doctor reported mesh had eroded into my bladder, attached to my bowels, my nerves and my muscles,” said Jodie Callahan, a mother who had surgery in 2010 at the age of 45.
Three years later, and following years of pain, she had the mesh removed. But recently learned she has to undergo another surgery after she said her doctor found more plastic mesh inside her body.
“This is affecting lives and it's major and no one is really listening to us,” said Barbara Melling.
Former IT manager for the FDA, Madris Tomes, was at that hearing too.
“I wanted to present data on all types of mesh, but they only wanted to see it for stress urinary incontinence or pelvic organ prolapse,” said Tomes.
Tomes left the FDA and started Device Events LLC, which runs a website that makes information about problems with medical devices easy to find and understand.
“I did a search on hernia mesh and I found 25,000 adverse event reports to the FDA and over 200 of those reports were deaths,” said Tomes.
FDA order How did we get here?
Last month the FDA ordered manufacturers to stop selling pelvic mesh. Yet made no mention of hernia mesh, stating that “The FDA has determined that the manufacturers, Boston Scientific and Coloplast, have not demonstrated a reasonable assurance of safety and effectiveness for these devices, which is the pre-market review standard that now applies to them since the agency reclassified them in class III (high risk) in 2016.”
“They really need to be looking at what are all the other devices just like this,” said Tomes.
"None of these products have gone through the rigorous process of being FDA approved. They're all cleared,” said Nafash.
She’s referring to the 510(k) process, which the KVUE Defenders reported on in March.
“If you look at the clearance process and look at the application to see in the application the company is telling the FDA this product is safe and effective because it is substantially similar to these other products already on the market. But if you look at the products they are claiming to be substantially similar to, those are also products that were cleared," she said. "And if you look at those products, those products were cleared based on other products that were cleared. If you continue down the line, it’s very dangerous. Often, products that aren’t even used in the same region as the product that is sought to be cleared, are what the product is claimed to be substantially similar. There are different forces at work in those regions, different organs and layers of tissue and muscle. I think it is the companies themselves that are responsible for putting a product on the market that is safe."
Why do surgeons use mesh? A doctor's perspective
Dr. Vineet Choudhry does not treat Pacheco but is a general surgeon specializing in hernia repair.
“I've done close to 1,000 hernia repairs and put in mesh in almost every single one of them,” said Dr. Choudhry.
He said, years ago, hernias were repaired using sutures or biologic mesh made from animal tissue or human cells.
“That mesh tends to absorb away over time, gets real soft and pliable and doesn't last very long. So, it's not really a permanent solution, but it helps temporarily,” he said.
Plastic mesh seemed to last longer.
A National Institutes of Health report shows the 10-year incisional hernia recurrence rate is reported to be 63% for traditional suture repair without mesh and 32% for repairs using prosthetic mesh.
It's been used by surgeons since the 1950s, but more frequently since the 1980s.
“The great majority of patients tolerate the mesh extremely well and don't even know that it's in there,” said Dr. Choudhry.
He said he has only had to remove one mesh implant from a patient who suffered an allergic reaction.
“When mesh goes well, it goes really well, but when it doesn't, it's not just a little bit of pain, it's disabling – where people need multiple surgeries – as many as six surgeries, 10 surgeries,” said Tomes.
“The mesh doesn't stay where it's put by itself, [it has to be sutured in place] over a period of time it will incorporate into the surrounding structure, but as it's healing it can definitely shift or move,” said Choudhry.
“These meshes are intended as permanent implants. Hernia mesh is used to resolve that hernia. Due to design defects in the meshes, often there is a surgery to remove that mesh. That mesh is sometimes adhered to the bowel or the omentum [the fatty tissue that covers and supports the intestines]. In some cases, people who have had these meshes have to have portions of their bowel or omentum or other internal organs removed,” said Nafash.
The problems tend to be reported 10 years or longer after the initial surgery. Device Events shows the problems increasing especially in the last two years.
The most common ones include "infection," mesh that adheres to internal organs like the colon or bowel even sepsis and death.
“I have bowel obstructions,” said Pacheco. “I’m throwing up bile every morning.”
Pacheco is not alone. Device Events found more than 4,700 bowel obstructions more than 5,500 infections – all people with hernia mesh – most happening since 2007.
“I had a hernia the size of a cherry, now they tell me it's the size of a tortilla. It's huge,” said Pacheco.
She used to weigh 225 pounds. Now she is barely 125 and facing another surgery by an out-of-state doctor who said her only hope is to add more mesh.
“They destroyed my life – the mesh,” she said.
Why no class action?
Medical device cases are typically MDL or multidistrict litigation cases. They are different than a class action.
“In a class action, you have people who have been injured in the same way. In mass tort litigation, while they have been injured by the same products, no person really is injured in the same way,” said Nafash.
In mass tort, each case is individually filed with the intention of having only a few cases go to trial.
The spinal cord stimulator A Marine is left paralyzed
From years to living with back pain to a life-changing procedure. Only for one New Braunfels veteran, the change was not what he expected.
“I did three tours in Okinawa. I did a tour down in Guantanamo Bay before 9/11,” said Winnett.
David Winnett Jr. retired from the Marines as a Captain after spending 20 years and countless tours around the world. The training took its toll.
“I developed degenerative discs. I started developing severe back pain the last two to three years of my career,” he said.
For years, pain pills helped. Then doctors pushed David to consider an alternative – a spinal cord stimulator.
“They told me I could relieve the back pain and get off the opiates,” he said.
Stimulators use electrical currents to block pain signals before they reach the brain. Each year, 50,000 are implanted worldwide.
He scheduled surgery for September 2017, a one-week trial to see if he should get the permanent implant.
“I came out of recovery and, within about a minute of waking up, I felt a very intense pain in my left groin,” he recalled.
He stood up for what he did not realize would be the last time.
“I laid back down and the exact same thing began happening on the other side, intense pain and then the pain hit my back. It was like someone was stabbing me in the spine. It was just horrible I just started screaming,” Winnett said. “Around that same time my legs went to sleep.”
Doctors flew David to Austin for emergency surgery removing the stimulator first.
“I thought the pain at the clinic in my groin was bad. Oh no, this was like a thousand times worse,” he said. “The next day I woke up in ICU. The doctor who put the device in came to visit me and he was apologizing me and I said, ‘Hey doc, that pain, when they took that thing out, was horrendous.’ And he said, ‘Didn't they turn it off?’ He looks at me. And I think, why didn't he turn it off?”
He took a video while recovering in the hospital that showed the lights were still flashing, the power to the stimulator was still on.
“It literally felt like I had sparks inside my spine it was that bad,” he said. “An MRI taken right after they pulled them out in Austin showed that one of the leads had taken a wrong turn. It hadn't gone straight in.”
Doctors determined the stimulator hit a blood vessel causing blood to fill his spinal column killing the nerves of his spine. The life-changing surgery changed him in ways he didn't imagine.
“I'm a paraplegic. This has just been a nightmare,” he said.
Winnett is not alone. Device Events.com found 681 people have been paralyzed by spinal cord stimulators. And more than 100,000 other injuries – most since 2008.
Attorney Dan Ross has represented a number of patients with medical device injuries.
“There are many heartbreaking stories, including David’s. Companies have been successful in getting judges to throw out these cases because they are preempted by a law that doesn't offer any remedies,” said Ross.
The device David had implanted was approved a month before his surgery through the pre-market approval (PMA) process, meaning it did undergo some human clinical trials.
People harmed, killed or disabled by a medical device that the U.S. Food and Drug Administration classifies as Class III cannot sue the manufacturer. Class III devices have the highest risk – they include cardiac stents, pacemakers and stimulators.
This is due to a 2008 U.S. Supreme Court ruling. The case Riegel vs. Medtronic granted “preemption” protection to makers of devices that undergo PMA. The court ruled because the devices undergo clinical trials and studies they are subject to liability protection.
That means Winnett cannot sue for what happened. It’s a loophole Congressman Lloyd Doggett and others want to fix with the medical device safety act.
"It's important that doctors get this information in and that we have the right. Everyone has the right to seek redress for their injuries and that these companies don't hide behind the FDA,” said Doggett.
It is too late for David.
“I did not plan to spend my retirement this way,” said Winnett. “Ironically even after this paraplegia, I still have the same back pain because I'm paralyzed below that level.”
He makes the best of each day sharing his story in hopes of helping others know the risks.
“I tried to do the right thing, what's politically correct nowadays, to get off the opiates well look what happened to me as a result. If I'd had it to do over I would have slept in that day,” he said.

The hidden reports Millions of problems not publically available
For decades the FDA has allowed certain medical device manufacturers to report problems with devices in papers that only government workers could see. Problems that remained hidden from patients, doctors and you.
These hidden reports were designed to hold information about known side effects for medical devices – things that were not thought to be dangerous.
But the KVUE Defenders uncovered some of those reports were hiding issues like cancer.
Life for Lacey Heatherly has never been clearer. It's been five months since a surgeon removed her breast implants.
“A week after I lost so much inflammation. Within seven days I'd lost seven pounds, my eyes got clearer, my skin got clearer, my brain,” she said.
Heatherly’s symptoms were similar to thousands of other women, but she had no way of knowing.
“The FDA has millions and millions of reports that are not publicly available,” said Madris Tomes, former IT manager for the FDA, who started Device Events LLC.
She said for decades the FDA has allowed certain manufacturers to file known side effects in what is known as alternative summary reports (ASRs), a name later changed to Voluntary Malfunction Summary Reporting (VMSR) program.
“The FDA gives them [device manufacturers] an exemption and says you can submit these in summation. So what that is, is a spreadsheet and it's not publicly available. It is sent to the FDA and they file it in storage, and if you are trying to find the true number of problems with a device you can't see those reports. You don't even know they exist. So you don't even know what to ask for,” said Tomes.
This program requires manufacturers to report deaths or serious injury events. The system was set up in the 1970s as a way to make reporting problems easier for medical device manufacturers, but Madris discovered one report could contain thousands of problems.
They had become a way to hide the true scope of what may be happening with certain devices. Case in point – breast implants.
About two years ago I had discovered summary reports were being used for breast implants. I did not know the extent of it, but what I'd found was that device companies thought a rupture wasn't a serious injury, and so they were saying we can put this in a summary report,” said Tomes.
When she also found cases of lymphoma being hidden in these summary reports, she brought her findings to the FDA.
“It took my finding this pattern for the FDA to say wait maybe these shouldn't be coming in as summaries,” she said.
Two years later, the FDA ordered manufacturers to make those issues public. Problems jumped from 51,000 to nearly 300,000 overnight.
"So 90% of the reports that existed the public didn't know about until that day,” Tomes said.
The FDA then ordered hearings. Dozens of women and doctors testified.
“How many lives have to be lost before action takes place? Isn't one life enough,” Jennifer Rob questioned the FDA panel at a hearing in February.
“After doing my research and finding out that the FDA and plastic surgeons knew prior to 2015 surgery that textured implants could cause this lymphoma, I feel sad and angry,” said Roxanne Vermiland.
More than just breast implants
It's not just breast implants. Several manufacturers including those that make implantable cardiac defibrillators, pacemakers, mesh and tooth implants, have been allowed to file these summary reports for decades. A practice the FDA announced it is ending.
“This is part of a larger effort to end the alternative summary reporting program for all medical devices, which we intend to complete in the coming weeks.”
But not before countless people like Heatherly suffered silently.
“I'm so glad I got these things out of my body,” said Heatherly.
Her experience has changed the way she views her own health.
“It makes me weary of the FDA, period,” she said. “Are you completely trusting the doctor says and what the FDA says? If you think this just affects breast implants, you are so wrong because this concerns everything we are doing for our bodies.”
Lymphoma by device Doctor will no longer implant devices that triggered patient's cancer
Since 2011, 457 women were treated for lymphoma, a rare cancer doctors say was caused by textured breast implants. Nine women died.
One of those women lives in Dripping Springs. Her case triggered her doctor to take extreme action.
Doctor visits take on a whole new meaning when you've had cancer. Especially a rare cancer you should have never had.
Sandra Rush nearly died from lymphoma. A cancer doctors say she developed from her textured breast implants.
“I was in a lot of pain,” she said.
Lymphoma was a cancer most doctors didn't know was linked to textured breast implants because the cases were hidden in summary reports that, until months ago, only the FDA could see.
“I never had heard about it. Nothing,” said Rush.
Dr. Elisabeth Potter had heard the cancer was rare.
“When I was training, I learned there was a cancer that could happen to women who had breast implants placed, but I heard it was very rare – one in a million. I never expected to see it in my own practice,” said Dr. Potter.
Dr. Potter helped reconstruct Rush’s breast tissue after she removed her textured breast implants. As she does with all surgeries, she removed more of the tissue surrounding the breast implant and sent it to a lab to be sure all the cancer was out.
RELATED:
"It was at that surgery that I found the real surprise," she said.
The test results showed Rush's lymphoma had spread.
"I sent what should have been healthy tissue to pathology and, when I got the pathology report back after reconstruction, it showed that the cancer had invaded into her breast. I was shocked," she said. "This cancer is not supposed to be a breast cancer and it’s not supposed to invade into the breast tissue. This breast implant associated cancer, which had occurred as the result of a cosmetic implant, was now in her bones in the rest of her body.”
“Two weeks in and I started feeling pain in my jaw, in the mandible. She [Dr. Potter] sent me in for a PET scan and that’s where they found it had metastasized all through my body,” Rush said.
Rush needed five rounds of heavy chemotherapy and a stem cell transplant, treatment that left her in the hospital for months.
“It was really, really difficult,” she said.
Rush was not the only patient with textured breast implants that Dr. Potter had seen develop lymphoma. She had seen two cases while in school. Rush was now the third. Too many, she said, for something not to change.
“In my mind, it is not safe enough to place a textured implant at this time. I have personally removed them from my practice," said Dr. Potter. "We tell every woman Sandra [Rush's[ story. The risk of that cancer is between one in 3,000 and one in 38,000. It is far from one in a million."

Other countries ban textured breast implants
Textured breast implants have been banned in Europe and Canada. Following a series of hearings this year, the FDA has decided to further study the issue.
"I hoped the FDA would simply remove the textured implant from the market, while we found out more details. They chose not to do so. Unfortunately, those implants are still being placed," she said.
Twenty-five years ago, Rush expected those implants to change her life, just not in this way.
“It changes your world it changes your life. It definitely does,” she said.
Dr. Potter said there are options for women looking to use their own tissue for breast reconstruction or augmentation work. As part of Diep flap reconstruction, surgeons use fat from a woman’s belly and then implant that in the breast area. This is now a common surgery Dr. Potter performs at her practice.
Efforts to change the laws Medica device safety acts put in place
Traveling is not easy for Frances Scott, a result of a metal-on-metal hip implant that she said left her poisoned and her muscles damaged.
She shared her story with KVUE and met with Congressman Lloyd Doggett for the first time in May. Doggett wanted to meet with Frances because he is pushing for two bills to increase transparency of medical device problems.
The Medical Device Safety Act will allow people to sue medical device manufacturers, even if they have gone through the PMA process to get the device cleared.
The Medical Device Guardian Act would require physicians and physician’s offices to report adverse events to the FDA. Right now, there is no requirement. Only hospitals and medical device manufacturers are required to report adverse events to the FDA.
For good reason.
According to Device Events, a service set up by a former FDA worker to better track problems with medical devices, the FDA receives more than 80,000 reports of problems each month involving various devices.
“The FDA takes two months to two years to identify a problem with a device,” said Tomes. “It makes me feel like the FDA is failing patients and failing physicians. We’re putting our healthcare system at risk because the FDA is not acting.”
That antiquated system that makes it difficult for patients and doctors to understand the true scope of problems.
“I think we need legislation to mandate a registry. Some within the FDA think a registry is a substitute for effective pre-approval testing I don’t. I think we need them both,” said Congressman Doggett. “We need that information to be out there so that everyone can be fully informed before they take the big step of an implant.”
Similar bills have failed to pass several times, but there is bipartisan support this time. And the FDA announced it will release summary reports on medical devices, which were previously hidden from the public.
If you have had experiences with medical device dangers, we encourage you to join the Medical Device Dangers Facebook group.