
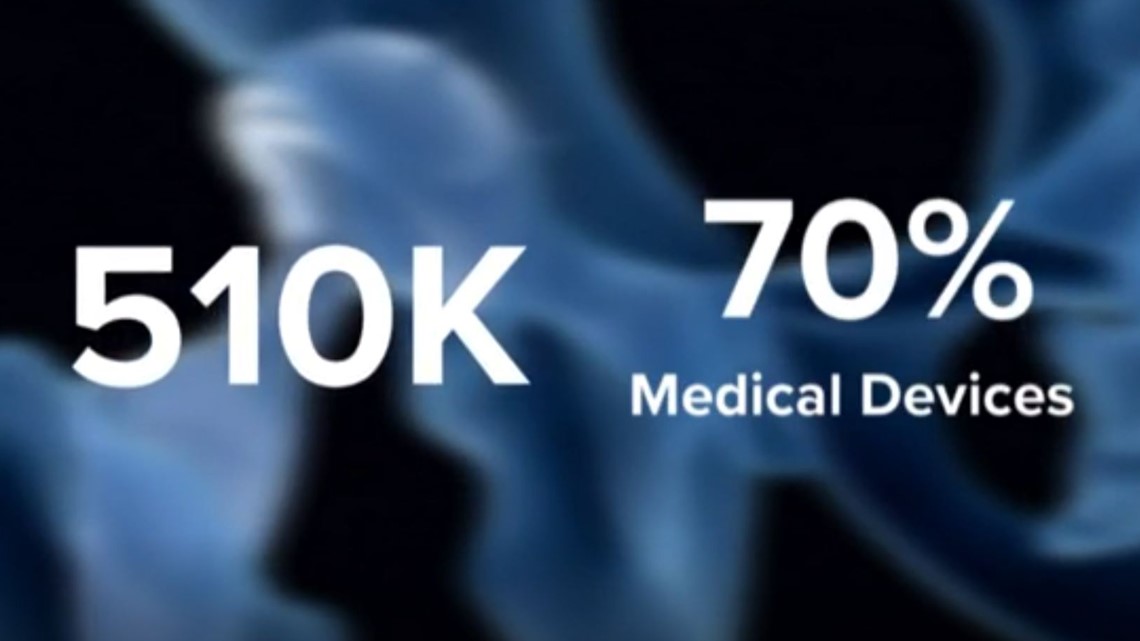
AUSTIN, Texas — Back in the 1970s, a process created to help encourage innovation is now how 70 percent of medical devices get approved.
“It's mind blowing. It’s just really upsetting,” said Frances Scott.
Scott spends a lot of time each day connecting with other people online.
“I know there are a lot of victims out there because they are calling me,” she said.
Scott shared her story with the KVUE Defenders after suffering for years from metal poisoning following a double hip replacement. A metal-on-metal hip that did not get tested in humans before it was allowed on the market by the FDA.
“It's a loophole,” said attorney Mark Lanier.
Lanier does not represent Frances but represents hundreds of people suing the makers of metal-on-metal hips.
“So many of the doctors that we've put under oath about this were stunned to find out that the company never tested the device in people,” said Lanier.
Devices are categorized first by class: Class I, II and III. According to the FDA, “Regulatory control increases from Class I to Class III. The device classification regulation defines the regulatory requirements for a general device type. Most Class I devices are exempt from Pre-Market Notification 510(k); most Class II devices require Pre-Market Notification 510(k); and most Class III devices require Pre-Market Approval.
A description of device classification and a link to the Product Classification Database is available here.
Devices like hip replacements, pacemakers and stimulators can be approved in two ways: Pre-Market Approval, or PMA, and 510(k).
PMA is the most stringent. Manufacturers must provide scientific evidence that the device is safe and effective, and most of the time this involves human trials.
510(k) is a process the FDA enacted back in the 1970s as a way to get devices that have had some type of improvement on the market faster.
Those devices only had to prove that they were substantially equivalent to a device already on the market.
The problem, as The New England Journal of Medicine pointed out in 2013, is that some of the comparable devices had been "voluntarily recalled."
“The problem is the companies figured out how to exploit that, in what the new England Journal of Medicine called a ‘loophole,’” said Lanier.
In the last five years, 70 percent of the medical devices approved in the U.S. were approved through the 510(k) process, meaning out of the majority of devices approved, more than 18,000 were not tested in people.

“This whole thing really saddens me,” said Scott.
It is why Scott is grateful Congressman Lloyd Doggett plans to push for a medical device registry.
“We need legislation and we need to demand accountability,” said Congressman Doggett.
“I think we need legislation to mandate a registry. Some within the FDA think a registry is a substitute for effective pre-approval testing. I don't. I think we need them both,” he said.
“Until we get it, we're not safe,” said Scott.
She knows the need. Even today she's lucky to get two hours a day where she is not in pain while standing. So she spends a lot of time online answering emails from others who continue to suffer.
Congressman Doggett's Medical Device Safety Act is expected to be submitted next month.
The FDA recognizes the need for changes to the 510(k) process and has begun implementing some.
It is requiring manufacturers of devices like metal-on-metal hips, and even breast implants and mesh, to show scientific proof that their products are safe. But only after thousands of people reported injuries.
The reporting of injuries is one of the many obstacles to finding out the dangers of certain medical devices. The KVUE Defenders will have more on that story in the coming months.
If you have had experiences with medical device dangers, we encourage you to join the Medical Device Dangers Facebook group.